|
This is the GENOA file server at MIT, providing access to genome alignments that detect loci
and pertinent alternative transcript structures of genes in genomic sequences for the human genome |
|
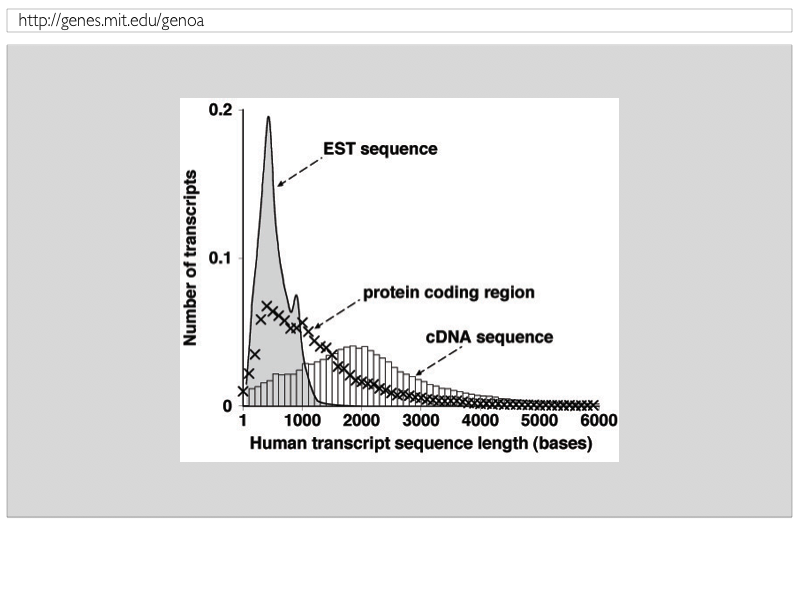
Figure1: Length distribution of spliced-aligned cDNAs, protein-coding regions and EST sequences |
| Transcripts for complementary DNA (cDNA), annotated protein-coding (CDS) regions for cDNAs, and expressed sequence tag (EST) sequences were obtained from GenBank, and splice-aligned to human genomic DNA. Figure 1shows the empirical length distribution of cDNA, CDS, and EST sequences. While ESTs are comparatively short at centered around 600 bases, cDNAs are centered around 2,000 bases with annotated CDS regions of average length of about 1,000 bases. |
|
|
|
Figure 2: Distribution of statictically significant BLAST searches of cDNAs against genomic DNA |
| For each human chromosome, interspersed repeat-masked cDNAs (rm-cDNAs) are searched against all loci of transcribed gene regions, and the best statistically significant BLAST rm-cDNA to genomic hit is retained for downstream spliced-alignments. Figure 2 shows the distribution of BLAST-aligned rm-cDNAs to genomic DNA, and it is found that the majority of rm-cDNAs is found to be BLAST-aligned to a single locus, while about 15,000 have multiple BLAST hits to different loci. The insert fo Fig.2 enlarges the number of multiple loci. |

|
|
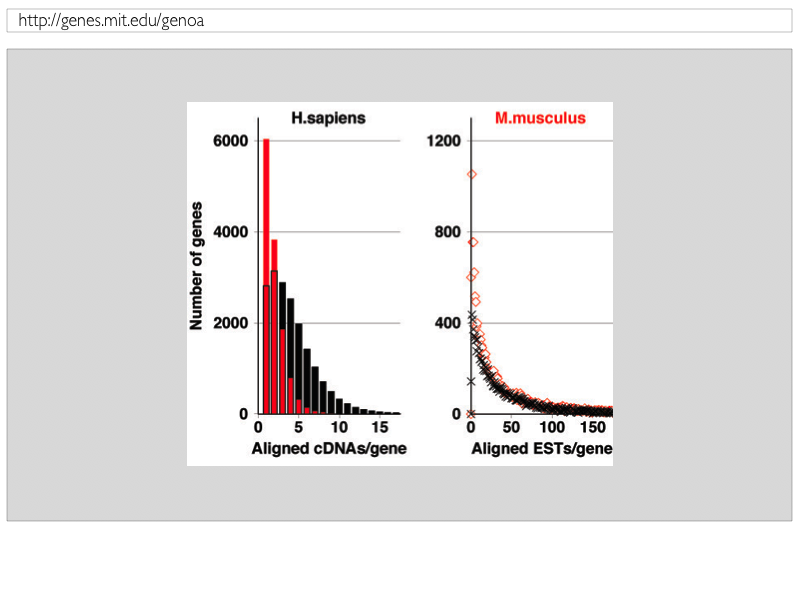
Figure 3: Transcript coverage of cDNA and EST sequences across gene loci |
| The average transcript coverage of human and mouse gene loci by spliced-aligned cDNA and EST sequences. Figure 3 shows that GENOA splice-aligns on average three to four human cDNAs for a typical gene locus, while the average number of cDNAs is smaller (one to two) for mouse genes. In comparison, the number of spliced-aligned ESTs is similar across both genomes. Figure 3 shows that both human and mouse genes show a large variation on EST transcript coverage, ranging from no to more than hundred ESTs, with similar distributions in both genomes. |
|
|
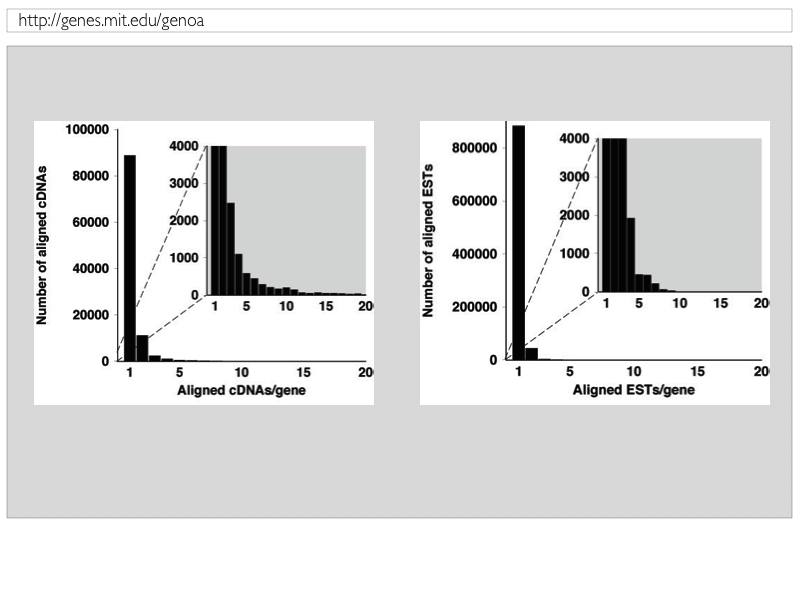
Figure 4: Distribution of spliced-alignments of cDNAs and ESTs against genomic DNA |
| Original cDNAs with statistically significant rm-cDNA BLAST searches are spliced-aligned against corresponding gene loci. The majority of cDNA is splice-aligned against a single locus (cf. Fig. 2). Figure 4 shows on the left hand side the distribution of splice-aligned cDNAs to genomic DNA. It is found that almost all cDNAs with statistically significant BLAST searches were determined to have spliced-alignments. It is further found that most cDNAs are splice-aligned to a single gene locus, while about 10,000 cDNAs were aligned to two and about 5,000 cDNAs to more than two loci (rapidly decaying for more than 15 loci) possibly reflecting paralogous genes with loci on the same chromosome. EST sequences were searched for statistically significant BLAST hits against rm-cDNAs with corresponding successfully obtained spliced-alignments, and the best hits were retained for donwstream spliced-alignments of ESTs against the corresponding genes. Figure 4 shows on the right hand side that the majority of ESTs is found to be splice-aligned to a single locus, while about 5,000 have multiple alignmnets to different loci. The inserts fo Fig.4 enlarges the number of multiple loci for both cDNA and EST sequences, respectively. |
|
|
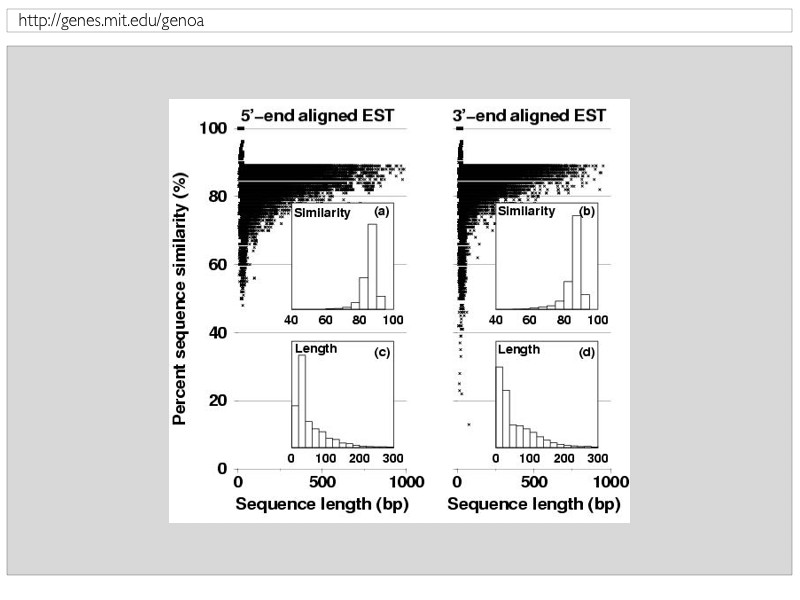
Figure 5: EST sequence size and percent identity of splice-aligned 5' and 3'-end sequence segments |
| Alignments of EST sequences to genomic DNA can be distorted due to spliced-alignments of small transcript segments of ESTs to genomically distant sequence regions. While potentially biologically meaningfull, spliced-alignmends of sizes such as about 20-30 bases were often found to be of spurious nature and would give rise to many skipped exons with "low-confidence" in the detected splicing event. To this end and in order to circumvent donstream effects, obtained spliced-alignments of ESts are further evaluated and subjected to the following criteria: (1) both the most 5'-end and 3'-end of each spliced-alignmend has to be equivalent or larger than 30 bases; (2) both the most 5'-end and 3'-end of each spliced-alignmend has to be equivalent or higher than 90% sequence similarity; (3) the whole spliced-alignmend has to be both equivalent or larger 90% in aligned sequence length compared to the orgiginal EST and to be equivalent or larger 90% in sequence similarity compared to the genomic DNA. Figure 5 shows the obtained distribution of sequence length and similarity for 5'-end and 3'-end EST sequence segments, respectively, as well the overall EST parameter. |
|
|
Please address comments/questions/suggestions regarding this webpage to Dirk Holste or Chris Burge Copyright © Chris Burge Lab, MIT, 2005 |